Cino EA, Choy WY, Karttunen M (2012)
Comparison of Secondary Structure Formation Using 10 Different Force Fields in
Microsecond Molecular Dynamics Simulations. J Chem Theory Comput 8:2725-2740. link to manuscript
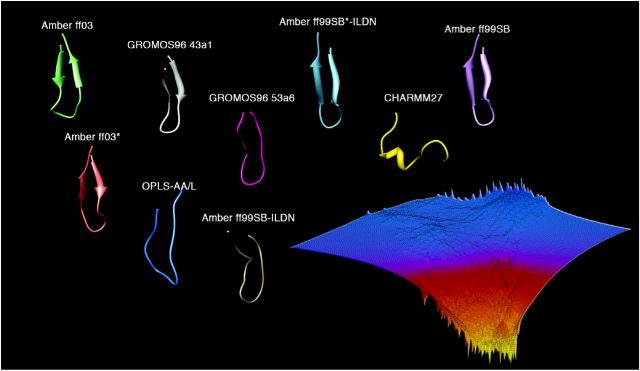
Fig 1. Structures of the NRF2 hairpin from folding simulations and
representative free energy landscape of the hairpin folding. The free
energy landscape was constructed from a 3 dimensional histogram consisting of radius
of gyration, backbone rmsd to bound state structure (PDB id: 2FLU) and distance
between 2 hydrophobic residues on opposite strands of the hairpin that make
close contacts (as determined by solution NMR for the peptide in the free
state).
A primary choice
in performing MD simulations is which force field to use. Currently, specific
force fields are employed depending on the system being investigated. For
example, a certain force field may give good agreement with experimental data
for a specific type of protein, but not necessarily for another. Even though
modifications to biomolecular force fields have lead to improved transferability,
further progress relies on continued testing. Ideally, these efforts will lead
to the development of fully transferable force fields.
A good method to test force field performance
is by simulating protein folding and comparing the results to experimentally
determined protein structures. However, most proteins fold on timescales unattainable
by modern computer simulations. As a result, it can be challenging to find good
test systems. One approach has been to extract amino acid sequences encoding self-folding
motifs out of well-folded proteins. While this may be a viable approach to
decrease system sizes and obtain folding events, care must be taken to ensure that
the motif does indeed fold properly in the absence of the rest of the protein. Another
approach has been to design small, fast folding proteins. However, protein
design is not an easy task.
Perhaps a better, in terms of being doable,
approach for force field testing of protein folding is to use amino acid
sequences encoding preformed structural elements (PSEs). As discussed in my January 11th post, intrinsically
disordered proteins (IDPs) often contain PSEs to facilitate their
interactions with other proteins. The benefits of using PSEs for folding
simulations is that they are typically locally occurring features that do not
rely as heavily upon long-range contacts as structural elements in well-folded
proteins. Moreover, they often contain features that are found in well-folded
proteins, such as hydrophobic clusters and electrostatic interactions. In many
ways, PSEs can be though of as mini or micro proteins. These may be ideal
candidates for testing of force fields.

Fig 2. Example of a hairpin motif. Hairpins are composed of two antiparallel beta strands connected by a turn. They are common structural elements found in many proteins.
Our related work references
1.
Cino
EA, Choy WY, Karttunen M (2012) Comparison of Secondary Structure Formation
Using 10 Different Force Fields in Microsecond Molecular Dynamics Simulations. J
Chem Theory Comput 8:2725-2740. link to manuscript
2.
Cino
EA, Wong-Ekkabut J, Karttunen M, Choy WY (2011) Microsecond molecular dynamics
simulations of intrinsically disordered proteins involved in the oxidative
stress response. PLoS One 6:e27371. link to manuscript
3.
Cino EA, Karttunen M, Choy WY (2012) Effects of molecular crowding on the
dynamics of intrinsically disordered proteins. PLoS One 7:e49876. link to manuscript
4.
Cino E, Fan J, Yang D, Choy WY (2012) (1)H, (15)N and (13)C backbone resonance
assignments of the Kelch domain of mouse Keap1. Biomol NMR Assign. In press. link to manuscript
5. Khan H, Cino, EA, Brickenden A, Fan J, Yang D, Choy WY (2013)
Fuzzy Complex Formation between the Intrinsically Disordered Prothymosin α and the Kelch Domain of
Keap1 Involved in the Oxidative Stress Response. J Mol Biol. In press. link to manuscript







